超晶格材料原子尺度建模
前言
虽然异质结的建模攻略很多,但大都以Material Studio为主,且对象是表界面。对于周期性的超晶格材料的建模,特别是异质结情况,参考较少,这里我分享一下个人的经验。采用ASE进行建模。
异质结的两种材料接触时,哪两个面间的接触,是需要确定的,比如说,根据实验确定,又或者无实验时,根据晶格匹配度确定,尽量保证失配度较低。
这里,假设已经确定了,两个材料的晶胞要沿着z轴堆叠。
整体思路:
1.确定好要合并的两个晶格的具体结构(用translate平移,surface切面,这个顺序好像也能反过来)
2.合并晶格(stack用起来还是蛮需要经验的)
如何切一个晶面,并生成周期性结构
from ase.build import surface,stack,make_supercell
from ase.io import read,write
from ase.io.vasp import write_vasp
BiTe = read('POSCAR_BiTe')
BiTe = make_supercell(BiTe,P=[[2,0,0],[0,1,0],[0,0,1]])
BiTe1 = BiTe.copy()
BiTe2 = BiTe.copy()
BiTe2.translate([0,0,-2.05985])
BiTe3 = BiTe.copy()
BiTe3.translate([0,0,-3.79553])
BiTe1 = surface(BiTe1,indices=(0,0,1),layers=1,periodic=True)
BiTe2 = surface(BiTe2,indices=(0,0,1),layers=1,periodic=True)
BiTe3 = surface(BiTe3,indices=(0,0,1),layers=1,periodic=True)
write_vasp('POSCAR_BiTe1',BiTe1,direct=True,sort=True)
write_vasp('POSCAR_BiTe2',BiTe2,direct=True,sort=True)
write_vasp('POSCAR_BiTe3',BiTe3,direct=True,sort=True)
ase的surface函数可以很简单的实现,这里不赘述了。
此外,关于如何确定一个合适的新晶格的大小,也有很多视频讲解,不再赘述。
改变晶体结构的原子层顺序
即便是只有一种原子的晶体,其沿某个面的堆垛的时候,可能有不同的层。比如FCC晶体沿001面的堆垛方式就是—ABAB—,显然与别的物质形成异质结时,会面临一个问题,即是A面还是B面与别的物质接触。
我们建模时,需要把A面或B面调整出来。这需要对原子进行整体位移
此外,异质结平面内的原子对齐(比如说xy面),也需要对原子进行整体位移。
而ASE实现原子整体位移非常简单,只需要用atoms类的translate方法即可,注意使用绝对坐标。
from ase.build import surface,stack,make_supercell
from ase.io import read,write
from ase.io.vasp import write_vasp
BiTe = read('POSCAR_BiTe')
BiTe = make_supercell(BiTe,P=[[2,0,0],[0,1,0],[0,0,1]])
BiTe1 = BiTe.copy()
BiTe2 = BiTe.copy()
BiTe2.translate([0,0,-2.05985])
BiTe3 = BiTe.copy()
BiTe3.translate([0,0,-3.79553])
ASE实现超晶格材料建模
from ase.build import surface,stack,make_supercell
from ase.io import read,write
from ase.io.vasp import write_vasp
BiTe1 = read('POSCAR_BiTe1')
BiTe2 = read('POSCAR_BiTe2')
BiTe3 = read('POSCAR_BiTe3')
Ag = make_supercell(read('POSCAR_Ag'),P=[[2,0,0],[0,2,0],[0,0,3]])
BiTe1_Ag = stack(Ag,BiTe1,axis=2,fix=0.5)
BiTe2_Ag = stack(Ag,BiTe2,axis=2,fix=0.5)
BiTe3_Ag = stack(Ag,BiTe3,axis=2,fix=0.5)
write('POSCAR_BiTe1_Ag',BiTe1_Ag,format='vasp')
write('POSCAR_BiTe2_Ag',BiTe2_Ag,format='vasp')
write('POSCAR_BiTe3_Ag',BiTe3_Ag,format='vasp')
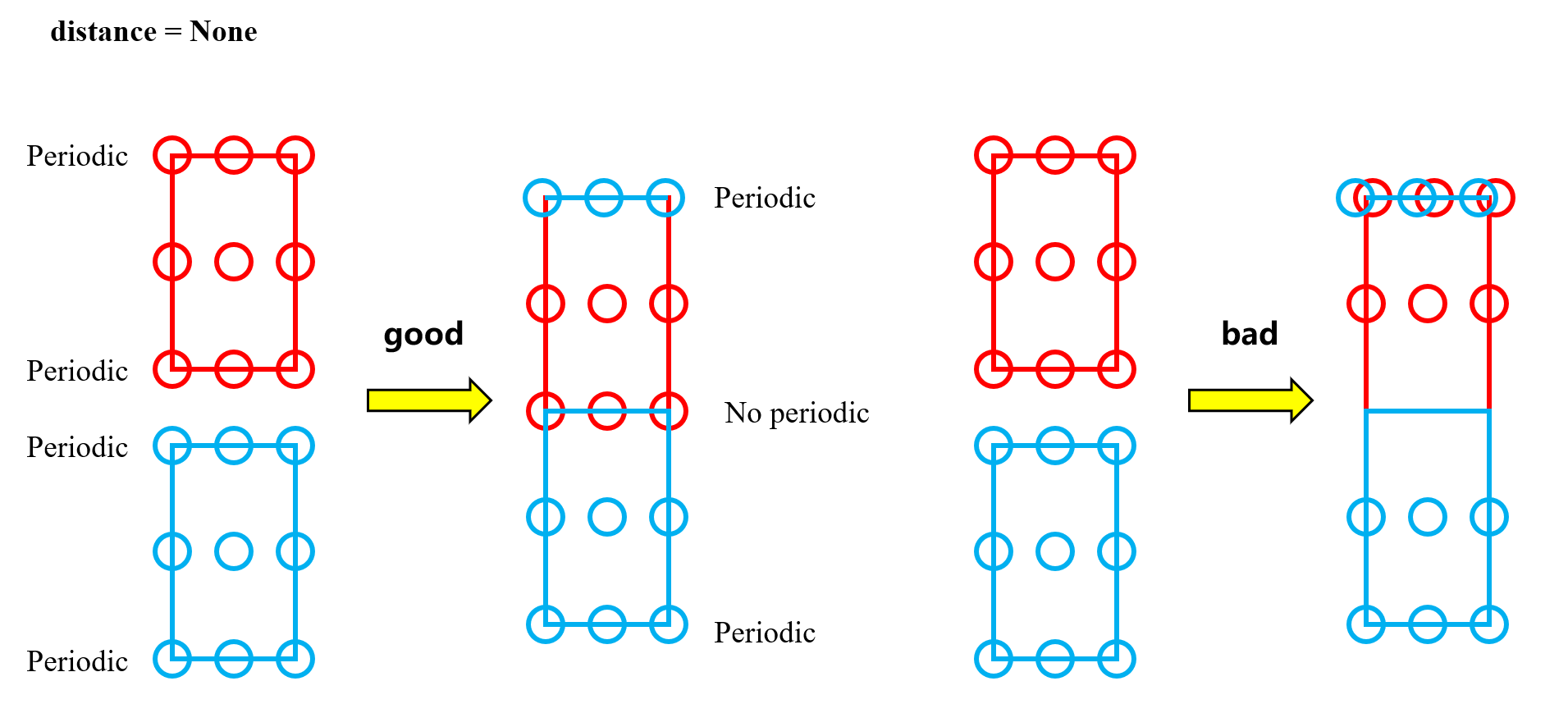
关于ase中stack函数的原理。
将两个周期性的晶胞合并成一个周期性的晶胞,有一个必须要面对的问题,那就是接触面将不再有周期性。
简单来讲,原本的四个周期性面,现在仅剩余两个。
然后,作为晶体结构文件的惯例,我们总是把原子放在周期性面上,即晶格边缘处,这样在显示时,会有重复显示的情况。
具体来讲,如图,红色格子和蓝色格子,其实只有两层原子,一层z坐标为0.0(分坐标),一层z为0.5,但显示时,z为1的情况也因为周期性显示出来了。
stack合并时,其实是把蓝色格子,(也就是第一个atoms对象),它的z=1的周期性面,保留到了最上方,即good情况。
那么什么是坏情况呢,坏情况是,红色格子的晶体结构文件中,两层原子坐标为z=0.0,z=1.0。
这种bad情况就会导致提到上方的红色原子与蓝色原子出现在同一平面上,不是我们期望的情况。
所以,请保证第二个atoms中,z=1.0或者几乎接近于1.0的原子,全部通过周期性换算成0,这很重要!
其实就是,两个晶胞的原子结构文件中,原子堆砌方向应该一致,不能一个从下堆砌到上,而另一个从上堆砌到下。
关于distance在控制些什么

distance其实是控制两个接触面间异质结原子的最小距离,会影响最终cell的z方向尺寸以及对原子进行微扰,所以最上和最下的两个面上的对称原子可能由于周期性,变成一层。