doped
用doped计算缺陷浓度随各种的变化!
Windows下运行的准备
因为doped会使用vise,而vise对Windows系统不是很友好,经过摸索,发现需要做两件事情才能让doped在Windows下顺利运行。
创建vise.yaml
经过测试,这个似乎是不必要的,但是我建议这么做。
在两个地方创建vise.yaml。
分别是c盘中,你的用户路径下,就是用来存放.condarc文件的路径。一般是:C:\Users\你的用户名\
你的python环境所在的盘的最浅路径下,比如说你的python在d盘,那么就应该是:D:\
vise.yaml的内容只需要是一个{}即可,不需要有真实内容。
修改vise软件包中的user_settings.py
他这个里的检索不怎么兼容Windows,导致我用doped,parse缺陷的时候一直卡住。
需要修改的内容,
# def _make_yaml_file_list(self) -> List[Path]:
# result = []
# dirname = self._cwd
# while True:
# filenames = [self.yaml_filename, "." + self.yaml_filename]
# file_paths = [dirname / filename for filename in filenames]
# for file_path in file_paths:
# if file_path.exists():
# result.append(file_path)
# if dirname == Path("/"):
# break
# else:
# dirname = dirname.parent
# return list(reversed(result))
把上面这个里的代码注释掉,(我已经注释过了)
换成新得,如下
def _make_yaml_file_list(self):
result = []
# 1. 当前目录
cwd_file = Path.cwd() / self.yaml_filename
if cwd_file.exists():
result.append(cwd_file)
# 2. 用户 home
home_file = Path.home() / self.yaml_filename
if home_file.exists():
result.append(home_file)
# 3. 可选环境变量指定
env_file = os.environ.get("VISE_YAML")
if env_file:
p = Path(env_file)
if p.exists():
result.append(p)
return result
doped所需的输入文件
可以根据doped来创建缺陷结构,但我接触doped比较晚,所以我是自己构建的缺陷结构并计算,所以只需要后处理就可以了。
parse规则
首先,我们创建一个用于存放,完美和缺陷结构计算结果的文件夹。比如说叫做Al2O3_Cr,这个名字是任意的。
然后在这个文件夹内,为每个缺陷结构和完美结构创建一个子文件夹。说起来比较抽象,直接看图吧
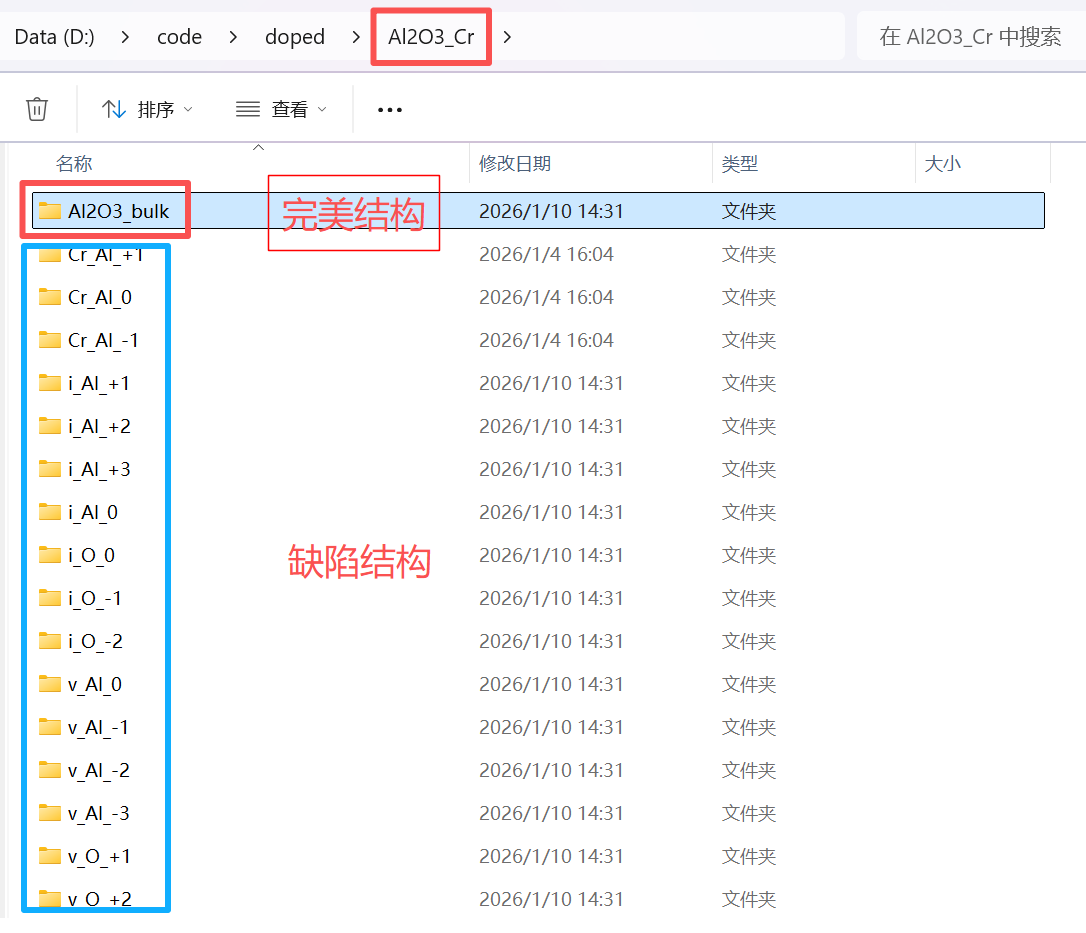
然后由于doped的内在逻辑,他会检索不同vasp版本的计算结果,所以这些文件夹内,不能直接放入vasprun.xml和OUTCAR。
Al2O3_bulk和Cr_Al_+1这些文件夹,要遵循一定的命名规则。
完美结构就是最后的后缀需要是,_bulk
而缺陷结构,则要,缺陷类型_占据格点类型_带有正负号的电荷量。
如Cr_Al_+1。
还要在缺陷和完美结构的文件夹内,创建一个名叫vasp_ncl的文件夹,然后放入vasprun.xml和OUTCAR。
具体如下:
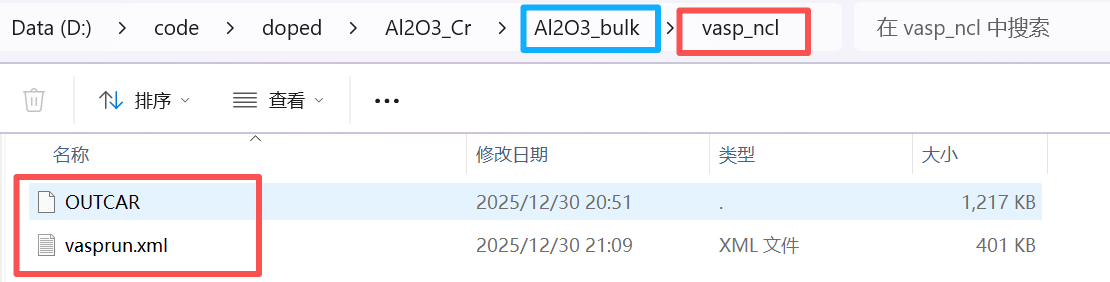
除了缺陷和完美结构,我们还需要一个路径来存放我们计算的dos,这个路径比较随意,也没有特别的命名规则,因为这个路径是直接在代码中指定的:
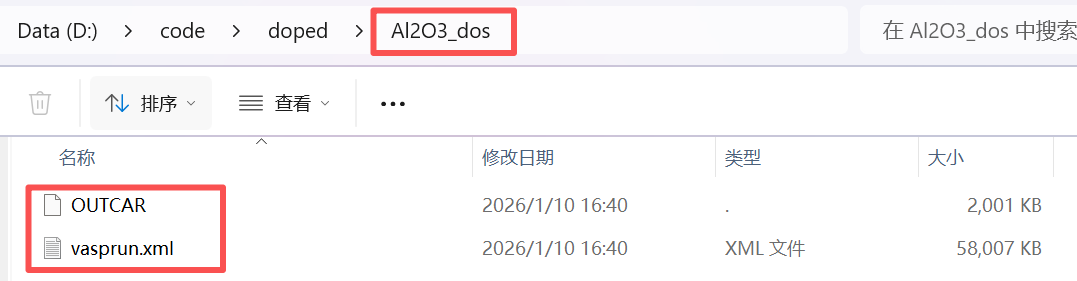
DOS的计算一定要用原胞!现在doped对非原胞的dos的处理存在问题!
DOS的计算一定要用原胞!现在doped对非原胞的dos的处理存在问题!
DOS的计算一定要用原胞!现在doped对非原胞的dos的处理存在问题!
化学势
对于刚接触doped的人来说,doped的化学势可能引人困惑。
虽然化学势可以parse得到,但我更推荐手写。(因为怎么parse的没看懂 = =)
简单来说,doped中定义了三种化学势,分别是绝对化学势,参考化学势(ref)和相对化学势(formal)。
绝对化学势,是DFT计算+修正项。
参考化学势是纯元素的DFT计算。
相对化学势是前两者之差。
参考化学势,比较好懂,就是DFT计算纯相,然后得到单个原子的能量。
绝对化学势呢
以下面这个化学势的写法为例。
Al2O3_chempots = {'limits': {'Opoor': {'O': -10.139, 'Al': -3.913}, 'Orich': {'O': -5.005, 'Al': -11.614}}, 'elemental_refs': {'O': -5.005, 'Al': -3.913}, 'limits_wrt_el_refs': {'Opoor': {'O': -5.134, 'Al': 0.0}, 'Orich': {'O': 0.0, 'Al': -7.701}}}
Orich的情况下,O的绝对化学势,就等于纯相的氧气分子总能的一半。
但是Al的绝对化学势,则是通过Al2O3和O2计算得到。
当然,O的绝对化学势也可以进一步进行有限温度和压强的修正。(如果修正了,Al的绝对化学势也会随之变动)
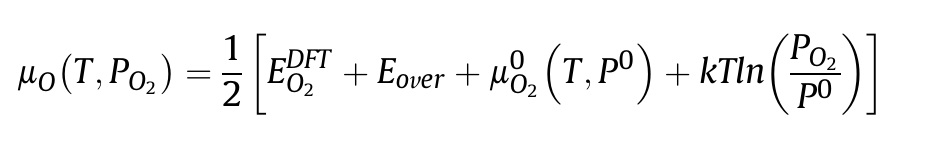
通过这个例子可以看到,绝对化学势除了和DFT计算有关(包括纯相O2和化合物A2O3的总能),和修正项也有关。
当然,除了doped格式的化学势字典,doped也支持最简单的写法。具体 参见手册
如果你只传入一种化学势,理论上,doped会把它视为绝对化学势。
但一旦在pase的环节,把相对化学势传递给doped后,他会变成这个实例的一个属性,doped会把后续传入的单一化学势都视为相对化学势。
所以,在这种情况下,我们后续传入单一化学势的时候,都需要先用绝对化学势和参考化学势做差,得到相对化学势再传入。
代码细节
pasing
from doped.analysis import DefectsParser
from monty.serialization import dumpfn, loadfn
dopedsys = 'Al2O3'
Al2O3_chempots = {'limits': {'Opoor': {'O': -10.139, 'Al': -3.913}, 'Orich': {'O': -5.005, 'Al': -11.614}}, 'elemental_refs': {'O': -5.005, 'Al': -3.913}, 'limits_wrt_el_refs': {'Opoor': {'O': -5.134, 'Al': 0.0}, 'Orich': {'O': 0.0, 'Al': -7.701}}}
dielectric = 9.13 # dielectric constant (this can be a single number (isotropic), or a 3x1 array or 3x3 matrix (anisotropic))
dp = DefectsParser("{}".format(dopedsys), processes=1, dielectric= dielectric, bulk_band_gap_vr ='Al2O3_dos/vasprun.xml') # dielectric needed for charge corrections
# Al2O3_chempots = {'O': -5.005, 'Al':-11.614}
Al2O3_thermo = dp.get_defect_thermodynamics(Al2O3_chempots)
dumpfn(Al2O3_thermo, fn="{}_thermo.json.gz".format(dopedsys))
这里化学势是用来处理缺陷形成能的,后续做热动力学部分,可以重新指定化学势。
比较重要的是,DefectsParser的processes=1,Windows多线程似乎容易出问题。
介电常数用于修正带电缺陷的形成能,对浓度计算影响比较大。
这里我传入化学势的时候,是用的doped格式的化学势字典,因此在dp这个实例中,储存下了ref的信息。
绘制缺陷浓度随气体分压变化的图
目标如下:
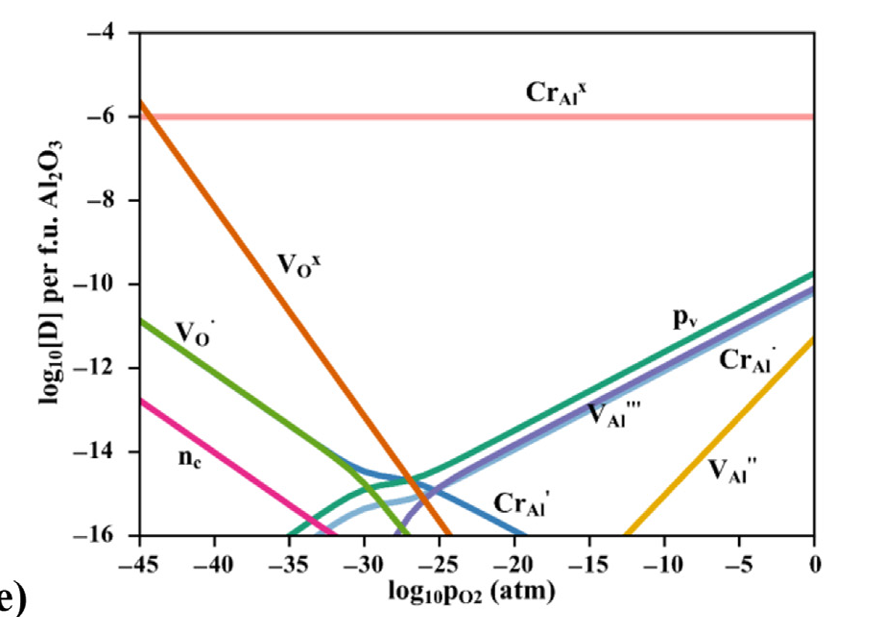
如前所述,气体的绝对化学势是受到分压和温度影响的。

我们可以通过查 热动力学表 来确定蓝色项,而红色项只和分压有关,所以氧的绝对化学势就变成了一个氧分压的函数。
所以我们要做的是生成一个化学势列表,这个列表里的一系列化学势,对应了一系列的氧分压(从小到1.0)。
代码如下:
import numpy as np
def chempots_at_x(x):
x_dict = {}
O_pot = -5.005 -2.542/2 + (np.log(np.power(10.0, np.array(x))) * 8.617 * 1e-5 * 1100)/2
x_dict['O'] = -2.542/2 + (np.log(np.power(10.0, np.array(x))) * 8.617 * 1e-5 * 1100)/2 +0.3
Al_pot = (-38.18 - 3 * O_pot)/2
x_dict['Al'] = Al_pot + 3.913
return x_dict
relative_chempots = []
pressure_indexs = np.linspace(-45, 0, 60)
for x in pressure_indexs:
relative_chempots.append(chempots_at_x(x))
print(relative_chempots)
你要注意以下几点:
- 我这里采用的是相对化学势,原因如前所述,我在解析DFT文件的环节,传入了参考化学势的值。
- -38.18是DFT计算的一个Al2O3分子的总能(我们假设温度和压强不影响固体的总能),+3.913是-(-3.913),即在求Al的相对化学势。
试着运行这段代码来看看它长什么样子!
创建一个FermiSolver的实例以及使用它的scan_chempots()方法
py_fs = FermiSolver(defect_thermodynamics=defect_thermo, chempots= Al2O3_chempots, bulk_dos='Al2O3_dos/vasprun.xml', backend="doped")
df = py_fs.scan_chempots(chempots=relative_chempots,temperature=1100,per_charge=True)
我想你注意到了,这里的化学势被传入了两次,第一次的无关紧要,其实和最初解析DFT文件时传入的是同一个。
而第二次在scan_chempots()中传入的,则是我们创建的一系列的化学势的列表。
此外,尽管目前doped的文档中声称只支持backend="py-sc-fermi",但其实已经支持了backend="doped"
我强烈建议使用backend="doped"!
然后你就会得到一个pandas定义的dataframe,虽然处理起来可能有些麻烦,但好在我想AI可以胜任接下来的工作。