AIMD
nvt系综
ISTART = 0
ENCUT = 480
PREC = Normal
ALGO = Fast
LREAL = Auto
LWAVE = .F.
LCHARG = .F.
GGA = PS
ISMEAR = 0
SIGMA = 0.05
ISPIN = 1
#electronstep
NELM = 200
NELMIN = 4
EDIFF = 1E-4
#AIMD
IBRION = 0
MDALGO = 2
ISIF = 2
TEBEG = 1500
NSW = 5000
POTIM = 0.5
SMASS = 0.5
IBRION是控制离子步优化算法的参数,进行分子动力学是设置为0。
MDALGO这个参数控制恒温器选项。
npt系综
ISTART = 0
ENCUT = 480
PREC = Normal
ALGO = Fast
LREAL = Auto
LWAVE = .F.
LCHARG = .F.
GGA = PS
ISMEAR = 0
SIGMA = 0.05
ISPIN = 1
#electronstep
NELM = 200
NELMIN = 4
EDIFF = 1E-4
#AIMD
IBRION = 0
MDALGO = 3
ISIF = 3
TEBEG = 1500
NSW = 5000
POTIM = 0.5
PMASS = 50
#O Zr Y H
LANGEVIN_GAMMA = 15 5 5 30
LANGEVIN_GAMMA_L = 1
MDALGO = 3使用拉格朗日热浴,支持npt系综。
PMASS控制
把OUTCAR压缩成.tar.gz文件
这个命令总是忘记, = =。
tar -czvf OUTCAR.tar.gz OUTCAR
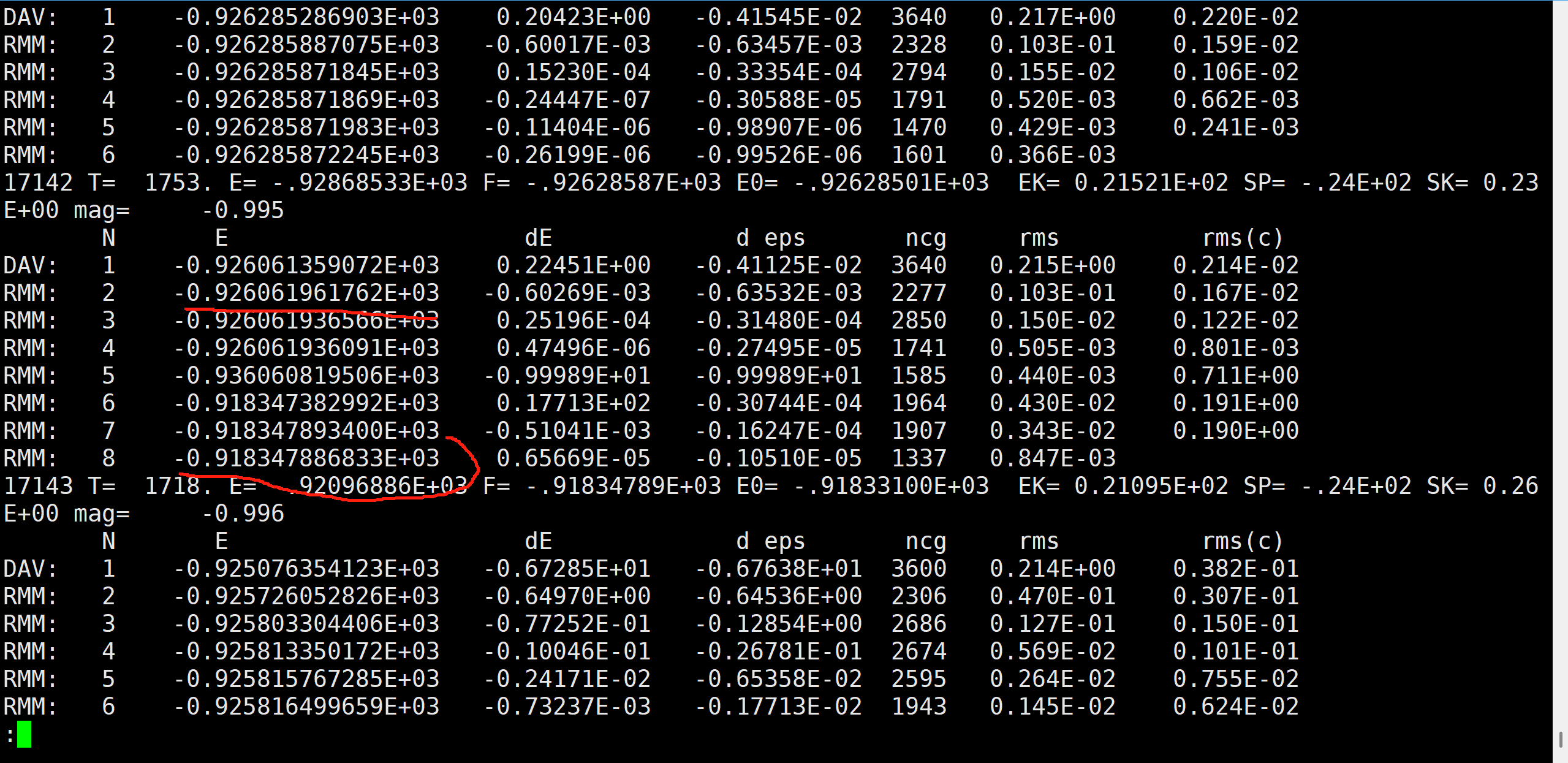
电子步收敛异常的现象
在进行AIMD的时候,出于某些未知的原因,一些构型在进行电子结构优化时,会陷入局部最小值或者未收敛。
具体表现为:1.达到了电子步的最大值(未收敛的情况),能量值与其他相似构型有明显差异(未收敛或陷入局部最小)。
这样的AIMD数据是坏的,不能进入ML-IAP的训练集。不然训练势的时候会有异常,如图:

可以看到,在拟合能量的时候(图3),有两个异常点,这是因为构型的电子结构计算没有收敛,得到的能量也是错误的。
当MACE开启第二阶段训练(加大能量权重)时,就会在损失函数上出现很严重的波动现象。
所以,在利用AIMD数据时,要进行一个检查,确保电子步没有达到最大值。
因为AIMD是一个采样加计算DFT的方法,所以个人觉得,有一两个构型出现不收敛,并不影响其他构型作为数据,把异常点剔除就行了。
检查OSZICAR
从OSZICAR中检查控温效果以及能量。
具体效果看注释。
from pymatgen.io.vasp.outputs import Oszicar
from matplotlib import pyplot as plt
import numpy as np
oszicar = Oszicar("YSZH/d3/d31/OSZICAR")
stepVStemp = []
stepVSenergy = []
#遍历轨迹的温度和能量
for trajectory, ionic_step in enumerate(oszicar.ionic_steps):
stepVStemp.append((trajectory,ionic_step['T']))
stepVSenergy.append((trajectory,ionic_step['F']))
# 用于打印是否能量有超出特定值
# if ionic_step['F'] > -920:
# print(trajectory)
#判断某一个轨迹的电子步是否达到最大,300是根据INCAR调整
for trajectory,electronic_step in enumerate(oszicar.electronic_steps):
if len(electronic_step) == 300:
print(trajectory)
print(len(electronic_step))
print("this AIMD data is not converged")
# 检查第二步电子步中能量与最后一步的变化是否大于5eV,可能是数据异常,这个判据好用,5eV根据体系调整
for trajectory,electronic_step in enumerate(oszicar.electronic_steps):
if abs(electronic_step[1]['E']-electronic_step[-1]['E']) > 5:
print(trajectory)
print('this AIMD data possibly is Local Minimum')
# 绘制温度与步数的关系
plt.figure()
#plt.plot(*zip(*stepVStemp), label='Temperature (K)')
plt.scatter(*zip(*stepVSenergy), label='Free Energy (eV)')
plt.xlabel('MD Step')
plt.ylabel('Value')
plt.legend()
plt.show()
# 曾用于电子步中检查dE确保收敛性,但不好用
#for trajectory,electronic_step in enumerate(oszicar.electronic_steps):
# electronic_step_bool_lsit = []
# for single_step in electronic_step:
# if abs(single_step['dE']) < 1E-5:
# electronic_step_bool = 1
# else:
# electronic_step_bool = 0
# electronic_step_bool_lsit.append(electronic_step_bool)
# is_non_decreasing = np.all(np.diff(electronic_step_bool_lsit) >= 0)
# if is_non_decreasing == False:
# print(trajectory)
# print("this AIMD data possibly is Local Minimum")
通过画图,可以看到AIMD中电子结构优化出现异常(未收敛或陷入局部最小)是比较常见的现象。尽管在一条轨迹中出现较少,但当轨迹数量增多时,难免会混入训练集中。
如何把他们提前筛选出来呢?
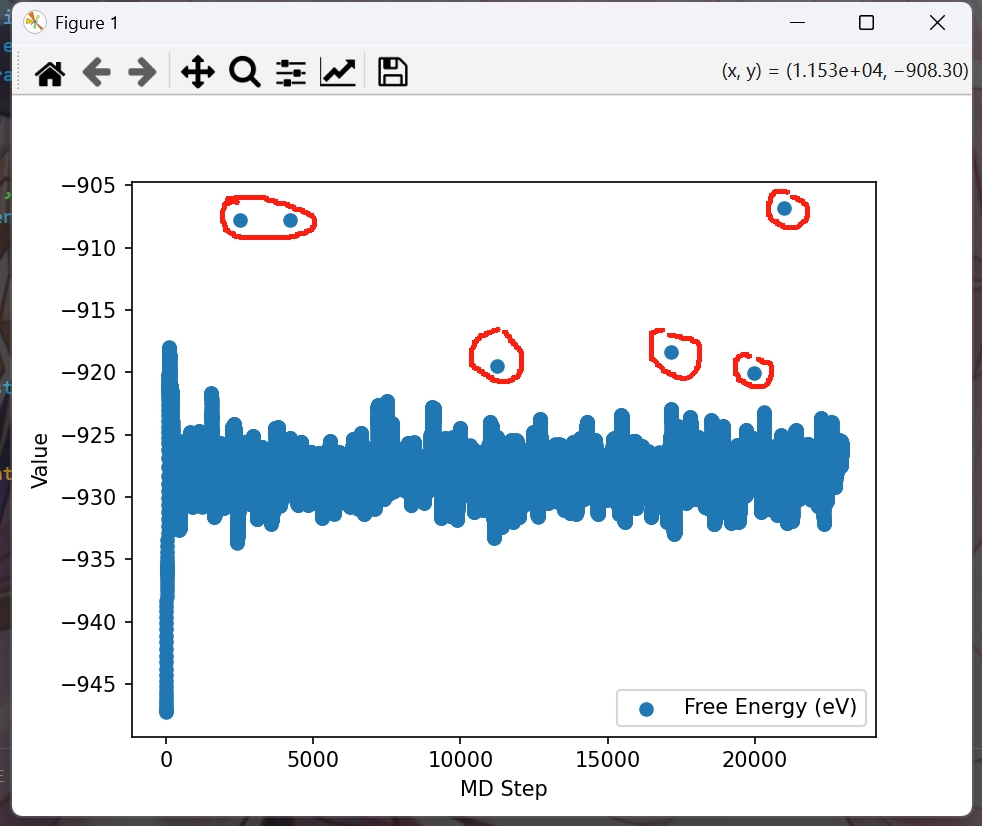
这里,可以检查某一构型对应的电子步是否达到最大,即检查是否收敛。
然后对于如何筛选陷入局部最小的结构,则是经验之谈。
如图,我发现对于陷入局部最优的结构,他们的能量变化,往往一开始是在全局最优波动,然后出于某些原因,来到了局部最优。
这样他们最初几个电子步的能量和最后的电子步能量差会比较大。对于我的结构,我用5eV作为门槛来判断。
发现效果不错。
所以检查AIMD的能量是非常有必要的。
用python脚本准备单点能的高通量计算
这个脚本被我放在了macetools文件夹下了(自我提醒用)
在训练机器学习势时,可能需要对一段轨迹重新进行单点能DFT计算。
这至少有两个应用背景:
- 用粗糙AIMD采样后得到的构型,需要重新进行高精度的DFT评估。
- 用ML-IAP进行MD后的构型,进行DFT评估,用于主动学习。
假设,这个脚本在一个路径下,这个路径放置了需要计算的轨迹(train.xyz)和这一批高通量计算所共用的INCAR、KPOINTS、POTCAR、以及提交任务的脚本(vasp.pbs)
我们先在这个路径下创建一个名为singlepoint的文件夹,然后运行脚本如下:
from ase.io import read
from ase.io.vasp import write_vasp
import os
import shutil
path = os.getcwd()
singlepoint_path = os.path.join(path,'singlepoint')
db = read('train.xyz',':')
for number,at in enumerate(db):
number_path = os.path.join(singlepoint_path,str(number))
os.makedirs(number_path)
#通用文件夹还是在超算上复制吧,自己电脑上复制后再上传太慢了
#INCAR_origin_path = os.path.join(path,'INCAR')
#INCAR_path = os.path.join(number_path,'INCAR')
#shutil.copy(INCAR_origin_path,INCAR_path)
#POTCAR_origin_path = os.path.join(path,'POTCAR')
#POTCAR_path =os.path.join(number_path,'POTCAR')
#shutil.copy(POTCAR_origin_path,POTCAR_path)
#KPOINTS_origin_path = os.path.join(path,'KPOINTS')
#KPOINTS_path = os.path.join(number_path,'KPOINTS')
#shutil.copy(KPOINTS_origin_path,KPOINTS_path)
#PBS_origin_path = os.path.join(path,'vasp.pbs')
#PBS_path = os.path.join(number_path,'vasp.pbs')
#shutil.copy(PBS_origin_path,PBS_path)
POSCAR_path = os.path.join(number_path,'POSCAR')
write_vasp(POSCAR_path,at,direct=True,sort=True)
单点能计算INCAR
#Start Parameters
PREC = N
ALGO = Fast
ISTART = 0
ICHARG = 2
GGA = PS
ISPIN = 1
LREAL = Auto
#Electronic Relaxation
NELM = 60
NELMIN = 4
EDIFF = 1E-5
LREAL = AUTO
ENCUT = 480
#Ionic Relaxation
NSW = 0
ISIF = 2
ISMEAR = 0
SIGMA = 0.05
#K
KSPACING = 0.5
经过测试,KSPACING=0.5和333的KPOINTS对于我的体系而言,计算得到的能量是差不多的,但是更快些。
ENCUT尝试等于600,但是时间翻了三倍,算了。
sh脚本提交批量提交超算任务
#!/bin/bash
# 提交 VASP 任务的循环脚本
# 文件夹名称从 0 到 199
for i in $(seq 0 199); do
echo "进入文件夹 $i"
cd "$i" || { echo "无法进入文件夹 $i"; exit 1; }
echo "提交任务:qsub vasp.pbs"
cp ../INCAR ./
cp ../KPOINTS ./
cp ../POTCAR ./
cp ../vasp.pbs ./
chmod +x vasp.pbs
qsub vasp.pbs
cd .. || exit
done
结构优化
# Start Parameters
PREC = N
ALGO = Fast
ISTART = 0
ICHARG = 2
GGA = PS
ISPIN = 1
LREAL = Auto
# Electronic Relaxation
NELM = 60
NELMIN = 4
EDIFF = 1E-5
ENCUT = 480
# Ionic Relaxation
NSW = 100
IBRION = 2
ISIF = 2
EDIFFG = -0.02 # 力收敛准则
ISMEAR = 0
SIGMA = 0.05
# K-points
KSPACING = 0.5