如何构建训练集用于训练机器学习势
前言
本文将介绍如何构建一个训练集,用于训练MACE势以及DP势。需要提前利用AIMD获取DFT数据集。这里的AIMD软件是VASP。
软件:ASE、DeepMD-Kit
注意:本文仅供参考,欢迎指出错误或分享补充。无能力提供任何指导,求教者切勿留言。
构建MACE势的训练集
MACE接受的训练集非常简单,一个xyz文件,包含了各种构型和它们对应的DFT数据标签,以及单原子的DFT数据,需要额外的标签config_type=IsolatedAtom。
顺带一提,ASE可以输出一种所谓的
Extended XYZ format
,会把各种各样的信息(有点类似OVITO中的全局信息),放到xyz文件的第二行。这一行会很长很长。MACE所采用的训练集输入格式就是它。
假设我们要构建水分子的MACE势,当前所处的路径下,有两个文件夹。
一个名为H2O的文件夹,里面存放着进行第一性原理分子动力学后得到的OUTCAR.tar.gz文件。路径为H2O/OUTCAR.tar.gz。
一个名为IsolatedAtoms的文件夹,里面存放着涉及元素(这里是H、O)的单原子的单点能计算(ISPIN=2)。路径分别为IsolatedAtoms/H/OUTCAR.tar.gz和IsolatedAtoms/O/OUTCAR.tar.gz。
代码展示
from ase.io import read,write
import random
#定义一个简单的函数用于打标签,这里可以自由更改标签的名字
def addlabel(configs,energy_label='energy_dft',forces_label='forces_dft',stress_label='stress_dft',is_isolated=False):
if is_isolated == False:
for at in configs:
at.info[energy_label] = at.get_potential_energy(force_consistent=True)
at.arrays[forces_label] = at.get_forces()
at.info[stress_label] = at.get_stress(voigt=True)
if is_isolated == True:
for at in configs:
at.info['config_type'] = 'IsolatedAtom'
at.info[energy_label] = at.get_potential_energy(force_consistent=True)
at.arrays[forces_label] = at.get_forces()
at.info[stress_label] = at.get_stress(voigt=True)
#read()函数,这里,第一个参数是所读文件路径,第二个参数是切片slice
IsolatedH = read('IsolatedAtoms/H/OUTCAR.tar.gz',':')
IsolatedO = read('IsolatedAtoms/O/OUTCAR.tar.gz',':')
IsolatedAtoms = IsolatedH + IsolatedO
addlabel(configs=IsolatedAtoms,is_isolated=True)
#这里的slice的意思是从第一个结构开始到最后一个结构,每100个结构取一个
db = read('H2O/OUTCAR.tar.gz','::100')
addlabel(configs=db)
#打乱训练集,这对训练非常重要
random.seed(42)
random.shuffle(db)
#将打过标签的数据集合并
db = db + IsolatedAtoms
write('trainset.xyz',db)
这里有趣的一点是,为什么对于单个结构的OUTCAR,也要进行切片:IsolatedH = read('IsolatedAtoms/H/OUTCAR.tar.gz',':'),而不是IsolatedH = read('IsolatedAtoms/H/OUTCAR.tar.gz')。
这是因为read()函数读取只有一个原子的结构是,会返回atom类的实例,而非atoms类的实例。而atom类不支持info属性,会很麻烦。
即便是多原子的单结构,如果想把他们合并成一个轨迹,也一定要用切片的形式":"读取,因为两个atoms类的实例加和会得到一个新的atoms类的实例。
from ase.io import read,write
#假设H2O.cif是一个含有一个水分子的胞
#这样只会输出一个含有两个水分子的胞
db1 = read('H2O.cif')
db2 = read('H2O.cif')
db = db1 + db2
write('H2O.xyz',db)
#使用切片后,read会返回list[atoms],再进行加和得到的是list[atoms1,atoms2],用write()函数写的时候,就能依次形成轨迹了
db1 = read('H2O.cif',':')
db2 = read('H2O.cif',':')
db = db1 + db2
write('H2O.xyz',db)
所以,如果我们想让单一结构也加入到我们的数据集时,也要记得用切片的形式进行读取。不过要铭记于心的是,用切片的形式读取的其实是一个list[atoms],要使用其中atoms实例的方法时,记得用for循环遍历其中的atoms实例。
拟合势的energy是vasp计算中的哪个energy?
虽然mace开发者已经指明,使用at.info[energy_label] = at.get_potential_energy(force_consistent=True)获取能量即可。
但还是好奇这个能量对应于OSZICAR中的哪个。
做了测试,对于单点能,OSZICAR包含F和E0两种能量,上述代码提取的是F,我的体系很多情况下F和E0是相等的。
对于AIMD,OSZICAR包含E、F、E0三种能量,第一个E其实是包含了动能,但我们拟合势只需要势能的值,所以提取的也还是F。
其实就是OUTCAR中的free energy TOTEN。
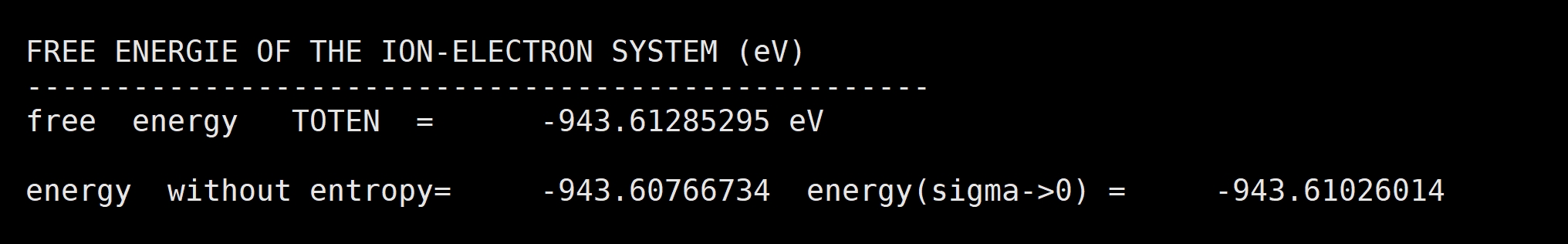
至于F和E0到底有什么区别?VASP没说明白,还需要更多实践。
AIMD的数据间隔取多少合适
个人认为这个和AIMD的timestep有关键,我的系统因为含氢,所以取得是0.5fs。如果步长比较大,间隔可以适当小一些。
取AIMD的间隔,用100和50做了尝试。
前者有5k个结构,后者有1w个结构。
前者的损失函数最终为0.049,后者为0.042。感觉差不多。